江蘇抗體藥物內毒素檢測法規要求
來源:
發布時間:2025-11-28
如何準備樣品進行內毒素檢測呢?測試前,需要根據樣品實際情況進行樣本前處理。大多數樣品只需要稀釋,使用內毒素檢測試劑盒進行測試即可。如果樣品有蛋白酶干擾并導致假陽性結果,建議對樣品稀釋并70°C加熱5-15分鐘進行熱滅活處理。如需要,可以對滅活樣品進行進一步稀釋后檢測。如果樣品可能含有受β-葡聚糖,建議使用抗增液。β-葡聚糖可能來自酵母和纖維素材料。如果樣品中因含有內毒素結合物而存在抑制,可以嘗試使用分散劑。
針對特殊樣本,rCR 在細菌內毒素檢測中的不干擾稀釋倍數(NID)比重組C因子(rFC)更低。江蘇抗體藥物內毒素檢測法規要求
湖州申科生物細菌內毒素工作標準品(CSE)源自大腸桿菌(E.coli O111:B4),經過提取精制獲得脂多糖,并以細菌內毒素國家標準品為基準標定效價,每支效價處于 10 - 100EU ,量值準確且可靠。在工業內毒素檢測中,該標準品應用廣且關鍵。首先,能用于鱟試劑靈敏度復核實驗,幫助檢測人員準確確認鱟試劑對細菌內毒素的敏感程度,保障檢測方法的有效性;其次,可開展干擾試驗,評估樣品中可能存在的干擾物質對檢測結果的影響,以便優化檢測流程和方法;再者,能充當各種陽性對照,為檢測過程提供參照,確保檢測結果的準確性與可靠性。這款標準品不僅適用于凝膠法鱟試劑系列實驗,在光度法鱟試劑系列實驗中同樣能發揮重要作用。使用時,只需參照中國藥典2025版第四部通則 1143 的相關介紹進行操作即可。憑借效價標定準確和適用性的優勢,該標準品為生物制品、化學藥品等各類樣品的細菌內毒素檢測提供了穩定且可靠的標準,助力企業和檢測機構嚴格把控產品質量,滿足法規要求。
廣東細菌內毒素檢測技術升級內毒素檢測的方法多樣性、多影響因素及實驗干擾,會導致自檢數據與廠家數據存在差異。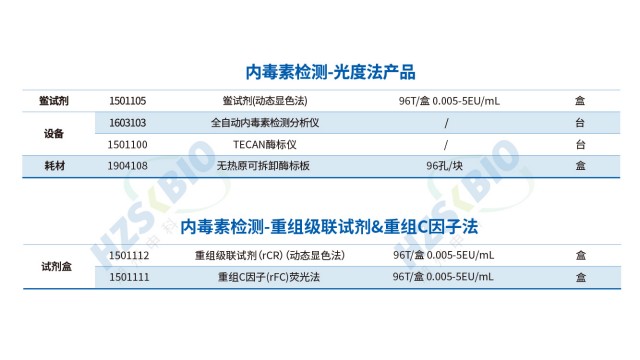
細菌內毒素是革蘭氏陰性菌細胞壁外膜釋放的脂多糖(LPS)成分,具有極強的生物活性,微量即可引發人體發熱、休克甚至多臟器功能衰竭。因此,在生物制品、醫療器械、制藥用水等領域,細菌內毒素檢測是保障產品安全性的關鍵質控環節。其檢測原理基于內毒素與特定試劑的特異性反應:內毒素可活化鱟血變形細胞裂解物(LAL)中的凝血級聯反應,通過C因子通路觸發酶促反應,通過觀察凝膠形成、濁度變化或顯色強度實現定量或定性分析。目前,各國藥典均將內毒素檢測列為強制要求,以降低臨床應用中的熱原風險。
鱟試驗法(LAL 法)是細菌內毒素檢測的經典方法,依據反應監測方式可分為三類:凝膠法、濁度法和顯色法。凝膠法通過觀察是否形成凝膠判斷內毒素是否超標,其優勢是操作簡便、成本較低,適用于定性或半定量檢測,如醫療器械初篩;濁度法通過監測反應體系濁度變化速率定量內毒素,靈敏度高(可達 0.005 EU/mL),適用于生物制品原液等高精度需求場景;顯色法基于顯色底物的吸光度變化定量,抗干擾能力較強,適配復雜基質樣品(如含蛋白質的注射液)。三種方法均需嚴格控制反應溫度(37℃±1℃)和時間,確保結果可靠性。
鱟試劑靈敏度復核至關重要,存放半年需重測,避免內毒素檢測出現假陰性或假陽性。
SHENTEK®凝膠法鱟試劑憑借多維度優勢,為細菌內毒素檢測提供高效解決方案: 法規契合度上,嚴格遵循中國藥典(ChP)、美國藥典(USP)、歐洲藥典(EP)及日本藥典(JP)的統一標準,確保檢測結果滿足全球藥品監管要求,為藥品研發、生產及出口提供合規支撐; 抗干擾性能上,配套申科特異性抗增液,可針對性抑制特殊樣品(如中藥注射劑、生物制品)的非特異性反應,突破復雜基質對檢測準確性的干擾,保障數據可靠; 性能適配性上,提供 0.03、0.06、0.125、0.25、0.5 EU/mL 多檔靈敏度選項,覆蓋從高靈敏到常規檢測的多樣化需求;操作易用性上,兼容恒溫儀、水浴鍋、恒溫箱等基礎設備,無需復雜儀器與軟件,降低實驗室硬件門檻,適配不同規模企業; 穩定性保障上,試劑采用凍干粉劑型,2~8℃ 儲存條件下有效期長達 2 年,減少倉儲與使用中的活性損耗,提升批次間一致性; 適用領域上,覆蓋生物制品、血液制品、化學藥品等全品類樣本,從工藝監控到成品放行,全流程支撐藥品安全質控。該試劑通過法規、性能、操作、穩定與適用的協同優化,構建起高效、靈活的內毒素檢測體系,助力行業提升質量管控水平。
動態顯色法鱟試劑監測吸光度變化定量內毒素,抗干擾強,適配復雜基質樣品檢測。合規性內毒素檢測技術服務細菌內毒素檢測中,rCR 對高蛋白(如單抗、FBS)、細胞培養上清等特殊樣本適配性好。江蘇抗體藥物內毒素檢測法規要求
低內毒素回收(LER)又稱內毒素掩蔽,是指無菌制劑(尤其蛋白類生物制劑)進行內毒素檢測時,加標內毒素的回收率<50% 的現象,且無法通過稀釋排除,區別于傳統檢測干擾。LER 會導致內毒素污染被低估,已被全球監管機構重點關注:FDA 2013 年要求生物藥 BLA 申報時提交 LER 研究報告;EMA 2023 年明確含表面活性劑(如吐溫)或螯合劑(如 EDTA)的制劑需提供 LER 數據;中國藥典 2025 版 9251 通則也新增 LER 內容,與國際接軌。這些要求促使企業優化內毒素檢測流程,避免因 LER 導致的檢測偏差,確保藥品安全。
江蘇抗體藥物內毒素檢測法規要求
下一篇:
非動物源內毒素檢測結果判定
相關新聞
- 北京重組蛋白熱原檢測MAT試劑盒 2025-12-09
- 北京細胞療法產品支原體檢測核酸擴增法 2025-12-09
- 吉林復雜基質支原體檢測核酸擴增法 2025-12-09
- 江蘇非動物源熱原檢測規范 2025-12-09
- 重慶疫苗產品支原體檢測技術服務 2025-12-09
- 安徽生物制品支原體檢測核酸擴增法 2025-12-09
- 安徽復雜基質支原體檢測技術服務 2025-12-09
- 山東細胞療法產品支原體檢測培養法 2025-12-09
- 合規性熱原檢測MAT試劑盒 2025-12-09
- 江蘇免疫細胞產品支原體檢測使用性驗證 2025-12-09
推薦新聞
- 湖北大鼠ELISA試劑盒電話多少 2025-12-09
- 上海黍峰生物鹽堿光合多通道冠層光合儀價錢 2025-12-09
- 長寧區比較好的內窺鏡服務費 2025-12-09
- 山西綜合高鹽核酸酶銷售電話 2025-12-09
- 虹口區是什么醫療管理服務規劃 2025-12-09
- 附近成人助聽器線下驗配地址 2025-12-09
- 成都數字化醫院病例查詢系統 2025-12-09
- 河北一體式供氧設備市價 2025-12-09
- 楊浦區服務易型EMA服務電話 2025-12-09
- 三文魚水光儀器售后服務 2025-12-09